Rosetta@home is a
distributed computingproject for
protein structure prediction on the
Berkeley Open Infrastructure for Network Computing (BOINC) platform, run by the
Baker laboratory at the
University of Washington. Rosetta@home aims to predict
protein–protein docking and
design new proteins with the help of about fifty-five thousand active volunteered computers processing at over 494,953 Giga
FLOPS on average as of March 26, 2020.
[5] Foldit, a Rosetta@Home videogame, aims to reach these goals with a
crowdsourcingapproach. Though much of the project is oriented toward basic
research to improve the accuracy and robustness of
proteomicsmethods, Rosetta@home also does applied research on
malaria,
Alzheimer's disease, and other pathologies.
[6]
Like all BOINC projects, Rosetta@home uses idle computer processing resources from volunteers' computers to perform calculations on individual
workunits. Completed results are sent to a central project
server where they are validated and assimilated into project
databases. The project is
cross-platform, and runs on a wide variety of hardware configurations. Users can view the progress of their individual
protein structure prediction on the Rosetta@home screensaver.
In addition to disease-related research, the Rosetta@home network serves as a testing framework for new methods in
structural bioinformatics. Such methods are then used in other Rosetta-based applications, like
RosettaDockor the
Human Proteome Folding Project and the
Microbiome Immunity Project, after being sufficiently developed and proven stable on Rosetta@home's large and diverse set of volunteer computers. Two especially important tests for the new methods developed in Rosetta@home are the
Critical Assessment of Techniques for Protein Structure Prediction (CASP) and
Critical Assessment of Prediction of Interactions (CAPRI) experiments, biennial experiments which evaluate the state of the art in protein structure prediction and protein–protein docking prediction, respectively. Rosetta@home consistently ranks among the foremost docking predictors, and is one of the best
tertiary structure predictors available.
[7]
With an influx of new users looking to participate in the fight against the
2019–20 coronavirus pandemic, caused by
SARS-CoV-2, Rosetta@home has increased its computing power up to 1.7 PetaFlops as of March 28, 2020.
[8][9]
Contents
1Computing platform
2Project significance
3Disease-related research
3.1Alzheimer's disease
3.2Anthrax
3.3Herpes simplex virus 1
3.4HIV
3.5Malaria
3.6Other diseases
4Development history and branches
4.1RosettaDesign
4.2RosettaDock
4.3Robetta
4.4Foldit
5Comparison to similar distributed computing projects
5.1Folding@home
5.2World Community Grid
5.3Predictor@home
6Volunteer contributions
7References
8External links
Computing platform[
edit]
See also:
List of distributed computing projects
The Rosetta@home application and the
BOINC distributed computing platform are available for the operating systems
Windows,
Linux, and
macOS; BOINC also runs on several others, e.g., FreeBSD.
[10] Participation in Rosetta@home requires a
central processing unit (CPU) with a
clock speed of at least 500
MHz, 200
megabytes of free
disk space, 512 megabytes of
physical memory, and Internet connectivity.
[11] As of July 20, 2016, the current version of the Rosetta Mini application is 3.73.
[12] The current recommended BOINC program version is 7.6.22.
[10] Standard
Hypertext Transfer Protocol (HTTP) (
port 80) is used for communication between the user's BOINC client and the Rosetta@home servers at the University of Washington;
HTTPS (port 443) is used during password exchange. Remote and local control of the BOINC client use port 31416 and port 1043, which might need to be specifically unblocked if they are behind a
firewall.
[13] Workunits containing data on individual proteins are distributed from servers located in the Baker lab at the
University of Washington to volunteers' computers, which then calculate a structure prediction for the assigned protein. To avoid duplicate structure predictions on a given protein, each workunit is initialized with a
random seed number. This gives each prediction a unique trajectory of descent along the protein's
energy landscape.
[14] Protein structure predictions from Rosetta@home are approximations of a
global minimum in a given protein's energy landscape. That global minimum represents the most energetically favorable conformation of the protein, i.e., its
native state.
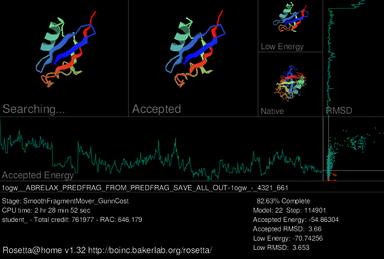
Rosetta@home
screensaver, showing the progress of a structure prediction for a synthetic
ubiquitin protein (PDB ID: 1ogw)
A primary feature of the Rosetta@home
graphical user interface (GUI) is a
screensaverwhich shows a current
workunit's progress during the simulated
protein foldingprocess. In the upper-left of the current screensaver, the target protein is shown adopting different shapes (conformations) in its search for the lowest energy structure. Depicted immediately to the right is the structure of the most recently accepted. On the upper right the lowest energy conformation of the current decoy is shown; below that is the true, or native, structure of the protein if it has already been determined. Three graphs are included in the screensaver. Near the middle, a graph for the accepted model's
thermodynamic free energy is displayed, which fluctuates as the accepted model changes. A graph of the accepted model's
root-mean-square deviation (RMSD), which measures how structurally similar the accepted model is to the native model, is shown far right. On the right of the accepted energy graph and below the RMSD graph, the results from these two functions are used to produce an energy vs. RMSD plot as the model is progressively refined.
[15]
Like all BOINC projects, Rosetta@home runs in the background of the user's computer, using idle computer power, either at or before logging into an account on the host
operating system. The program frees resources from the CPU as they are needed by other applications so that normal computer use is unaffected. Many program settings can be specified via user account preferences, including: the maximum percentage of CPU resources the program can use (to control power consumption or heat production from a computer running at sustained capacity), the times of day during which the program can run, and many more.
Rosetta, the software that runs on the Rosetta@home network, was rewritten in
C++ to allow easier development than that allowed by its original version, which was written in
Fortran. This new version is
object-oriented, and was released on February 8, 2008.
[12][16] Development of the Rosetta code is done by Rosetta Commons.
[17] The software is freely licensed to the academic community and available to pharmaceutical companies for a fee.
[17]
Project significance[
edit]
Further information:
Protein structure prediction,
Protein docking, and
Protein design
With the proliferation of
genome sequencing projects, scientists can infer the amino acid sequence, or
primary structure, of many proteins that carry out functions within the cell. To better understand a protein's function and aid in
rational drug design, scientists need to know the protein's three-dimensional
tertiary structure.
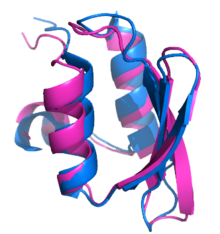
CASP6 target T0281, the first ab initio protein structure prediction to approach atomic-level resolution. Rosetta produced a model for T0281 (
superpositioned in magenta) 1.5
Ångström (Å)
RMSD from the crystal structure (blue).
Protein 3D structures are currently determined experimentally via
X-ray crystallography or
nuclear magnetic resonance (NMR) spectroscopy. The process is slow (it can take weeks or even months to figure out how to crystallize a protein for the first time) and costly (around US$100,000 per protein).
[18] Unfortunately, the rate at which new sequences are discovered far exceeds the rate of structure determination – out of more than 7,400,000 protein sequences available in the
National Center for Biotechnology Information (NCBI) nonredundant (nr) protein database, fewer than 52,000 proteins' 3D structures have been solved and deposited in the
Protein Data Bank, the main repository for structural information on proteins.
[19] One of the main goals of Rosetta@home is to predict protein structures with the same accuracy as existing methods, but in a way that requires significantly less time and money. Rosetta@home also develops methods to determine the structure and docking of
membrane proteins (e.g.,
G protein–coupled receptors (GPCRs)),
[20] which are exceptionally difficult to analyze with traditional techniques like X-ray crystallography and NMR spectroscopy, yet represent the majority of targets for modern drugs.
Progress in protein structure prediction is evaluated in the biannual
Critical Assessment of Techniques for Protein Structure Prediction (CASP) experiment, in which researchers from around the world attempt to derive a protein's structure from the protein's amino acid sequence. High scoring groups in this sometimes competitive experiment are considered the de facto standard-bearers for what is the state of the art in protein structure prediction. Rosetta, the program on which Rosetta@home is based, has been used since CASP5 in 2002. In the 2004 CASP6 experiment, Rosetta made history by being the first to produce a close to atomic-level resolution, ab initio
protein structure prediction in its submitted model for CASP target T0281.
[21] Ab initio modeling is considered an especially difficult category of protein structure prediction, as it does not use information from
structural homology and must rely on information from
sequence homology and modeling physical interactions within the protein. Rosetta@home has been used in CASP since 2006, where it was among the top predictors in every category of structure prediction in CASP7.
[22][23][24] These high quality predictions were enabled by the computing power made available by Rosetta@home volunteers.
[25] Increasing computing power allows Rosetta@home to sample more regions of
conformation space (the possible shapes a protein can assume), which, according to
Levinthal's paradox, is predicted to
increase exponentially with protein length.
Rosetta@home is also used in
protein–protein docking prediction, which determines the structure of multiple
complexed proteins, or
quaternary structure. This type of
protein interaction affects many cellular functions, including antigen–antibody and enzyme–inhibitor binding and cellular import and export. Determining these interactions is critical for
drug design. Rosetta is used in the
Critical Assessment of Prediction of Interactions (CAPRI) experiment, which evaluates the state of the protein docking field similar to how CASP gauges progress in protein structure prediction. The computing power made available by Rosetta@home's project volunteers has been cited as a major factor in Rosetta's performance in CAPRI, where its docking predictions have been among the most accurate and complete.
[26]
In early 2008, Rosetta was used to computationally design a protein with a function never before observed in nature.
[27] This was inspired in part by the retraction of a high-profile paper from 2004 which originally described the computational design of a protein with improved enzymatic activity relative to its natural form.
[28] The 2008
research paper from David Baker's group describing how the protein was made, which cited Rosetta@home for the computing resources it made available, represented an important
proof of concept for this protein design method.
[27] This type of protein design could have future applications in drug discovery,
green chemistry, and
bioremediation.
[27]
Disease-related research[
edit]
In addition to basic research in predicting protein structure, docking and design, Rosetta@home is also used in immediate disease-related research.
[29] Numerous minor research projects are described in David Baker's Rosetta@home journal.
[30] As of February 2014, information on recent publications and a short description of the work are being updated on the forum.
[31] The forum thread is no longer used since 2016, and news on the research can be found on the general news section of the project.
[32]
Alzheimer's disease[
edit]
A component of the Rosetta software suite, RosettaDesign, was used to accurately predict which regions of amyloidogenic proteins were most likely to make
amyloid-like fibrils.
[33] By taking hexapeptides (six amino acid-long fragments) of a protein of interest and selecting the lowest energy match to a structure similar to that of a known fibril forming hexapeptide, RosettaDesign was able to identify peptides twice as likely to form fibrils as are random proteins.
[34]Rosetta@home was used in the same study to predict structures for
amyloid beta, a fibril-forming protein that has been postulated to cause
Alzheimer's disease.
[35] Preliminary but as yet unpublished results have been produced on Rosetta-designed proteins that may prevent fibrils from forming, although it is unknown whether it can prevent the disease.
[36]
Anthrax[
edit]
Another component of Rosetta, RosettaDock,
[37][38][39] was used in conjunction with experimental methods to model interactions between three proteins—lethal factor (LF), edema factor (EF) and protective antigen (PA)—that make up
anthrax toxin. The computer model accurately predicted docking between LF and PA, helping to establish which
domains of the respective proteins are involved in the LF–PA complex. This insight was eventually used in research resulting in improved anthrax vaccines.
[40][41]
Herpes simplex virus 1[
edit]
RosettaDock was used to model docking between an
antibody (
immunoglobulin G) and a surface protein expressed by the cold sore virus,
herpes simplex virus 1 (HSV-1) which serves to degrade the antiviral antibody. The protein complex predicted by RosettaDock closely agreed with the especially difficult-to-obtain experimental models, leading researchers to conclude that the docking method has potential to address some of the problems that X-ray crystallography has with modeling protein–protein interfaces.
[42]
HIV[
edit]
As part of research funded by a $19.4 million grant by the
Bill & Melinda Gates Foundation,
[43]Rosetta@home has been used in designing multiple possible vaccines for human immunodeficiency virus (
HIV).
[44][45]
Malaria[
edit]
In research involved with the
Grand Challenges in Global Health initiative,
[46] Rosetta has been used to computationally design novel
homing endonuclease proteins, which could eradicate
Anopheles gambiae or otherwise render the mosquito unable to transmit
malaria.
[47] Being able to model and alter protein–DNA interactions specifically, like those of homing endonucleases, gives computational protein design methods like Rosetta an important role in
gene therapy (which includes possible cancer treatments).
[29][48]
Other diseases[
edit]
Rosetta@home researchers have designed an
IL-2 receptor agonist called Neoleukin-2/15 that does not interact with the alpha subunit of the receptor. Such immunity signal molecules are useful in cancer treatment. While the natural IL-2 suffers from toxicity due to an interaction with the alpha subunit, the designed protein is much safer, at least in animal models.
[49] Rosetta molecular modeling suite was recently used to accurately predict the atomic-scale structure of the
SARS-CoV-2 spike protein weeks before it could be measured in the lab.
[50]
Development history and branches[
edit]
Originally introduced by the Baker laboratory in 1998 as an ab initio approach to structure prediction,
[51] Rosetta has since branched into several development streams and distinct services. The Rosetta platform derives its name from the
Rosetta Stone, as it attempts to decipher the structural "meaning" of proteins' amino acid sequences.
[52] More than seven years after Rosetta's first appearance, the Rosetta@home project was released (i.e., announced as no longer
beta) on October 6, 2005.
[12] Many of the graduate students and other researchers involved in Rosetta's initial development have since moved to other universities and research institutions, and subsequently enhanced different parts of the Rosetta project.
RosettaDesign[
edit]

Superposition of Rosetta-designed model (red) for
Top7 onto its
X-raycrystal structure (blue, PDB ID: 1QYS)
RosettaDesign, a computing approach to protein design based on Rosetta, began in 2000 with a study in redesigning the folding pathway of
Protein G.
[53] In 2002 RosettaDesign was used to design
Top7, a 93-amino acid long
α/β protein that had an overall
fold never before recorded in nature. This new conformation was predicted by Rosetta to within 1.2
Å RMSD of the structure determined by
X-ray crystallography, representing an unusually accurate structure prediction.
[54] Rosetta and RosettaDesign earned widespread recognition by being the first to design and accurately predict the structure of a novel protein of such length, as reflected by the 2002 paper describing the dual approach prompting two positive letters in the journal
Science,
[55][56] and being cited by more than 240 other scientific articles.
[57] The visible product of that research,
Top7, was featured as the RCSB PDB's 'Molecule of the Month' in October 2006;
[58] a
superposition of the respective cores (residues 60–79) of its predicted and X-ray crystal structures are featured in the Rosetta@home logo.
[21]
Brian Kuhlman, a former postdoctoral associate in
David Baker's lab and now an associate professor at the
University of North Carolina, Chapel Hill,
[59] offers RosettaDesign as an online service.
[60]
RosettaDock[
edit]
RosettaDock was added to the Rosetta software suite during the first
CAPRI experiment in 2002 as the Baker laboratory's
algorithm for
protein–protein docking prediction.
[61] In that experiment, RosettaDock made a high-accuracy prediction for the docking between
streptococcal pyogenic exotoxin A and a
T cell-receptor β-chain, and a medium accuracy prediction for a complex between
porcine α-amylase and a
camelid antibody. While the RosettaDock method only made two acceptably accurate predictions out of seven possible, this was enough to rank it seventh out of nineteen prediction methods in the first CAPRI assessment.
[61]
Development of RosettaDock diverged into two branches for subsequent CAPRI rounds as Jeffrey Gray, who laid the groundwork for RosettaDock while at the
University of Washington, continued working on the method in his new position at
Johns Hopkins University. Members of the Baker laboratory further developed RosettaDock in Gray's absence. The two versions differed slightly in side-chain modeling, decoy selection and other areas.
[39][62] Despite these differences, both the Baker and Gray methods performed well in the second CAPRI assessment, placing fifth and seventh respectively out of 30 predictor groups.
[63] Jeffrey Gray's RosettaDock server is available as a free docking prediction service for non-commercial use.
[64]
In October 2006, RosettaDock was integrated into Rosetta@home. The method used a fast, crude docking model phase using only the
protein backbone. This was followed by a slow full-atom refinement phase in which the orientation of the two interacting proteins relative to each other, and side-chain interactions at the protein–protein interface, were simultaneously optimized to find the lowest energy conformation.
[65] The vastly increased computing power afforded by the Rosetta@home network, combined with revised fold-tree representations for backbone flexibility and
loop modeling, made RosettaDock sixth out of 63 prediction groups in the third CAPRI assessment.
[7][26]
Robetta[
edit]
The Robetta (Rosetta Beta) server is an automated protein structure prediction service offered by the Baker laboratory for non-commercial ab initio and comparative modeling.
[66] It has participated as an automated prediction server in the biannual
CASP experiments since CASP5 in 2002, performing among the best in the automated server prediction category.
[67] Robetta has since competed in CASP6 and 7, where it did better than average among both automated server and human predictor groups.
[24][68][69] It also participates in the
CAMEO3D continuous evaluation.
In modeling protein structure as of CASP6, Robetta first searches for structural homologs using
BLAST,
PSI-BLAST, and
3D-Jury, then parses the target sequence into its individual
domains, or independently folding units of proteins, by matching the sequence to structural families in the
Pfam database. Domains with structural homologs then follow a "template-based model" (i.e.,
homology modeling) protocol. Here, the Baker laboratory's in-house alignment program, K*sync, produces a group of sequence homologs, and each of these is modeled by the Rosetta de novo method to produce a decoy (possible structure). The final structure prediction is selected by taking the
lowest energy model as determined by a low-resolution Rosetta energy function. For domains that have no detected structural homologs, a de novo protocol is followed in which the lowest energy model from a set of generated decoys is selected as the final prediction. These domain predictions are then connected together to investigate inter-domain, tertiary-level interactions within the protein. Finally, side-chain contributions are modeled using a protocol for
Monte Carlo conformational search.
[70]
In CASP8, Robetta was augmented to use Rosetta's high resolution all-atom refinement method,
[71]the absence of which was cited as the main cause for Robetta being less accurate than the Rosetta@home network in CASP7.
[25] In CASP11, a way to predict the
protein contact map by co-evolution of residues in related proteins called GREMLIN was added, allowing for more de novofold successes.
[72]
Foldit[
edit]
See also:
Foldit
On May 9, 2008, after Rosetta@home users suggested an interactive version of the
distributed computing program, the Baker lab publicly released
Foldit, an online protein structure prediction game based on the Rosetta platform.
[73] As of September 25, 2008, Foldit had over 59,000 registered users.
[74] The game gives users a set of controls (for example, shake, wiggle, rebuild) to manipulate the
backbone and amino acid
side chains of the target protein into more energetically favorable conformations. Users can work on solutions individually as soloists or collectively as evolvers, accruing points under either category as they improve their structure predictions.
[75]
Comparison to similar distributed computing projects[
edit]
There are several distributed computed projects which have study areas similar to those of Rosetta@home, but differ in their research approach:
Folding@home[
edit]
Of all the major distributed computing projects involved in protein research,
Folding@home is the only one not using the
BOINC platform.
[76][77][78] Both Rosetta@home and Folding@home study protein misfolding diseases such as
Alzheimer's disease, but Folding@home does so much more exclusively.
[79][80] Folding@home almost exclusively uses all-atom
molecular dynamics models to understand how and why proteins fold (or potentially misfold, and subsequently aggregate to cause diseases).
[81][82] In other words, Folding@home's strength is modeling the process of protein folding, while Rosetta@home's strength is computing protein design and predicting protein structure and docking.
Some of Rosetta@home's results are used as the basis for some Folding@home projects. Rosetta provides the most likely structure, but it is not definite if that is the form the molecule takes or whether or not it is viable. Folding@home can then be used to verify Rosetta@home's results, and can provide added atomic-level information, and details of how the molecule changes shape.
[82][83]
The two projects also differ significantly in their computing power and host diversity. Averaging about 6,650 tera
FLOPS from a host base of
central processing units (CPUs),
graphics processing units (GPUs), and
PS3s,
[84] Folding@home has nearly 108 times more computing power than Rosetta@home.
[85]
World Community Grid[
edit]
Both Phase I and Phase II of the
Human Proteome Folding Project (HPF), a subproject of
World Community Grid, have used the Rosetta program to make structural and functional annotations of various
genomes.
[86][87] Although he now uses it to create databases for biologists,
Richard Bonneau, head scientist of the Human Proteome Folding Project, was active in the original development of Rosetta at David Baker's laboratory while obtaining his PhD.
[88] More information on the relationship between the HPF1, HPF2 and Rosetta@home can be found on Richard Bonneau's website.
[89]
Predictor@home[
edit]
Like Rosetta@home,
Predictor@home specialized in protein structure prediction.
[90] While Rosetta@home uses the Rosetta program for its structure prediction, Predictor@home used the dTASSER methodology.
[91] In 2009, Predictor@home shut down.
Other protein related distributed computing projects on
BOINC include
QMC@home,
Docking@home,
POEM@home,
SIMAP, and
TANPAKU. RALPH@home, the Rosetta@home
alpha project which tests new application versions, work units, and updates before they move on to Rosetta@home, runs on BOINC also.
[92]
Volunteer contributions[
edit]
Rosetta@home depends on computing power donated by individual project members for its research. As of March 28, 2020, about 53,000 users from 150 countries were active members of Rosetta@home, together contributing idle processor time from about 54,800 computers for a combined average performance of over 1.7 Peta
FLOPS.
[85][93]
 Bar chart
Bar chart showing cumulative credit per day for Rosetta@home over a 60-day period, indicating its computing power during the
CASP8 experiment
Users are granted
BOINC credits as a measure of their contribution. The credit granted for each workunit is the number of
decoys produced for that workunit multiplied by the average claimed credit for the decoys submitted by all computer hosts for that workunit. This custom system was designed to address significant differences between credit granted to users with the standard BOINC client and an optimized BOINC client, and credit differences between users running Rosetta@home on
Windows and
Linuxoperating systems.
[94] The amount of credit granted per second of CPU work is lower for Rosetta@home than most other BOINC projects.
[95]Rosetta@home is thirteenth out of over 40 BOINC projects in terms of total credit.
[96]
Rosetta@home users who predict protein structures submitted for the CASP experiment are acknowledged in scientific publications regarding their results.
[25] Users who predict the lowest energy structure for a given workunit are featured on the Rosetta@home
homepage as Predictor of the Day, along with any team of which they are a member.
[97] A User of the Day is chosen randomly each day to be on the homepage also, from among users who have made a Rosetta@home profile.
[98]
References[
edit]
^ "Rosetta@home License Agreement". Boinc.bakerlab.org. Archived from
the original on April 19, 2014. Retrieved April 18, 2014.
^ "Portfolio Highlight: Rosetta++ Software Suite". UW TechTransfer – Digital Ventures. Retrieved September 7, 2008.
^ "Rosetta@home".
^ "Rosetta@Home – Detailed stats | BOINCstats/BAM!".
^ "Rosette@home".
^ "What is Rosetta@home?". Rosetta@home forums. University of Washington. Archived from
the original on September 13, 2008. Retrieved September 7, 2008.
^
Jump up to: a b Lensink MF, Méndez R, Wodak SJ (December 2007). "Docking and scoring protein complexes: CAPRI 3rd Edition". Proteins. 69 (4): 704–18.
doi:
10.1002/prot.21804.
PMID 17918726.
^ "Rosetta@home - Server Status "TeraFLOPS estimate"". Rosetta@home. March 25, 2020. Retrieved March 25, 2020.
^ "Rosetta@home Rallies a Legion of Computers Against the Coronavirus". HPCWire. March 24, 2020. Retrieved March 25, 2020.
^
Jump up to: a b "Download BOINC client software". BOINC. University of California. 2008. Retrieved December 1, 2008.
^ "Rosetta@home: Recommended System Requirements". Rosetta@home. University of Washington. 2008. Archived from
the original on September 25, 2008. Retrieved October 7, 2008.
^
Jump up to: a b c "Rosetta@home: News archive". Rosetta@home. University of Washington. 2016. Retrieved July 20, 2016.
^ "Rosetta@home: FAQ (work in progress) (message 10910)". Rosetta@home forums. University of Washington. 2006. Retrieved October 7, 2008.
^ Kim DE (2005).
"Rosetta@home: Random Seed (message 3155)". Rosetta@home forums. University of Washington. Retrieved October 7, 2008.
^ "Rosetta@home: Quick guide to Rosetta and its graphics". Rosetta@home. University of Washington. 2007. Archived from
the original on September 24, 2008. Retrieved October 7, 2008.
^ Kim DE (2008).
"Rosetta@home: Problems with minirosetta version 1.+ (Message 51199)". Rosetta@home forums. University of Washington. Retrieved September 7, 2008.
^
Jump up to: a b "Rosetta Commons". RosettaCommons.org. 2008. Archived from
the original on September 15, 2008. Retrieved October 7, 2008.
^ Bourne PE, Helge W, eds. (2003). Structural Bioinformatics. Hoboken, NJ: Wiley-Liss.
ISBN 978-0-471-20199-1.
OCLC 50199108.
^ "Yearly Growth of Protein Structures". RCSB Protein Data Bank. 2008. Retrieved November 30,2008.
^ Baker D (2008).
"Rosetta@home: David Baker's Rosetta@home journal (message 55893)". Rosetta@home forums. University of Washington. Retrieved October 7, 2008.
^
Jump up to: a b "Rosetta@home: Research Overview". Rosetta@home. University of Washington. 2007. Archived from
the original on September 25, 2008. Retrieved October 7, 2008.
^ Kopp J, Bordoli L, Battey JN, Kiefer F, Schwede T (2007). "Assessment of CASP7 predictions for template-based modeling targets". Proteins. 69 Suppl 8: 38–56.
doi:
10.1002/prot.21753.
PMID 17894352.
^ Read RJ, Chavali G (2007). "Assessment of CASP7 predictions in the high accuracy template-based modeling category". Proteins. 69 Suppl 8: 27–37.
doi:
10.1002/prot.21662.
PMID 17894351.
^
Jump up to: a b Jauch R, Yeo HC, Kolatkar PR, Clarke ND (2007). "Assessment of CASP7 structure predictions for template free targets". Proteins. 69 Suppl 8: 57–67.
doi:
10.1002/prot.21771.
PMID 17894330.
^
Jump up to: a b c Das R, Qian B, Raman S, et al. (2007). "Structure prediction for CASP7 targets using extensive all-atom refinement with Rosetta@home". Proteins. 69 Suppl 8: 118–28.
doi:
10.1002/prot.21636.
PMID 17894356.
^
Jump up to: a b Wang C, Schueler-Furman O, Andre I, et al. (December 2007). "RosettaDock in CAPRI rounds 6–12". Proteins. 69 (4): 758–63.
doi:
10.1002/prot.21684.
PMID 17671979.
^
Jump up to: a b c Jiang L, Althoff EA, Clemente FR, et al. (March 2008).
"De novo computational design of retro-aldol enzymes". Science. 319 (5868): 1387–91.
Bibcode:
2008Sci...319.1387J.
doi:
10.1126/science.1152692.
PMC 3431203.
PMID 18323453.
^ Hayden EC (February 13, 2008). "Protein prize up for grabs after retraction". Nature.
doi:
10.1038/news.2008.569.
^
Jump up to: a b "Disease Related Research". Rosetta@home. University of Washington. 2008. Archived from
the original on September 23, 2008. Retrieved October 8, 2008.
^ Baker D (2008).
"Rosetta@home: David Baker's Rosetta@home journal". Rosetta@home forums. University of Washington. Retrieved September 7, 2008.
^ "Rosetta@home Research Updates". Boinc.bakerlab.org. Retrieved April 18, 2014.
^ "News archive". Rosetta@home. Retrieved May 10, 2019.
^ Kuhlman B, Baker D (September 2000).
"Native protein sequences are close to optimal for their structures". Proceedings of the National Academy of Sciences of the United States of America. 97(19): 10383–88.
Bibcode:
2000PNAS...9710383K.
doi:
10.1073/pnas.97.19.10383.
PMC 27033.
PMID 10984534.
^ Thompson MJ, Sievers SA, Karanicolas J, Ivanova MI, Baker D, Eisenberg D (March 2006).
"The 3D profile method for identifying fibril-forming segments of proteins". Proceedings of the National Academy of Sciences of the United States of America. 103 (11): 4074–78.
Bibcode:
2006PNAS..103.4074T.
doi:
10.1073/pnas.0511295103.
PMC 1449648.
PMID 16537487.
^ Bradley P.
"Rosetta@home forum: Amyloid fibril structure prediction". Rosetta@home forums. University of Washington. Retrieved September 7, 2008.
^ Baker D.
"Rosetta@home forum: Publications on R@H's Alzheimer's work? (message 54681)". Rosetta@home forums. University of Washington. Retrieved October 8, 2008.
^ Wang C, Schueler-Furman O, Baker D (May 2005).
"Improved side-chain modeling for protein–protein docking". Protein Science. 14 (5): 1328–39.
doi:
10.1110/ps.041222905.
PMC 2253276.
PMID 15802647.
^ Gray JJ, Moughon S, Wang C, et al. (August 2003). "Protein–protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations". Journal of Molecular Biology. 331 (1): 281–99.
doi:
10.1016/S0022-2836(03)00670-3.
PMID 12875852.
^
Jump up to: a b Schueler-Furman O, Wang C, Baker D (August 2005). "Progress in protein–protein docking: atomic resolution predictions in the CAPRI experiment using RosettaDock with an improved treatment of side-chain flexibility". Proteins. 60 (2): 187–94.
doi:
10.1002/prot.20556.
PMID 15981249.
^ Lacy DB, Lin HC, Melnyk RA, et al. (November 2005).
"A model of anthrax toxin lethal factor bound to protective antigen". Proceedings of the National Academy of Sciences of the United States of America. 102 (45): 16409–14.
Bibcode:
2005PNAS..10216409L.
doi:
10.1073/pnas.0508259102.
PMC 1283467.
PMID 16251269.
^ Albrecht MT, Li H, Williamson ED, et al. (November 2007).
"Human monoclonal antibodies against anthrax lethal factor and protective antigen act independently to protect against Bacillus anthracis infection and enhance endogenous immunity to anthrax". Infection and Immunity. 75 (11): 5425–33.
doi:
10.1128/IAI.00261-07.
PMC 2168292.
PMID 17646360.
^ Sprague ER, Wang C, Baker D, Bjorkman PJ (June 2006).
"Crystal structure of the HSV-1 Fc receptor bound to Fc reveals a mechanism for antibody bipolar bridging". PLOS Biology. 4 (6): e148.
doi:
10.1371/journal.pbio.0040148.
PMC 1450327.
PMID 16646632.
^ Paulson, Tom (July 19, 2006).
"Gates Foundation awards $287 million for HIV vaccine research". Seattle Post-Intelligencer. Retrieved September 7, 2008.
^ Liu Y, et al. (2007).
"Development of IgG1 b12 scaffolds and HIV-1 env-based outer domain immunogens capable of eliciting and detecting IgG1 b12-like antibodies" (PDF). Global HIV Vaccine Enterprise. Archived from
the original (PDF) on February 25, 2009. Retrieved September 28, 2008.
^ Baker D.
"David Baker's Rosetta@home journal archives (message 40756)". Rosetta@home forums. University of Washington. Retrieved September 7, 2008.
^ "Homing Endonuclease Genes: New Tools for Mosquito Population Engineering and Control". Grand Challenges in Global Health. Retrieved September 7, 2008.
^ Windbichler N, Papathanos PA, Catteruccia F, Ranson H, Burt A, Crisanti A (2007).
"Homing endonuclease mediated gene targeting in Anopheles gambiae cells and embryos". Nucleic Acids Research. 35 (17): 5922–33.
doi:
10.1093/nar/gkm632.
PMC 2034484.
PMID 17726053.
^ Ashworth J, Havranek JJ, Duarte CM, et al. (June 2006).
"Computational redesign of endonuclease DNA binding and cleavage specificity". Nature. 441 (7093): 656–59.
Bibcode:
2006Natur.441..656A.
doi:
10.1038/nature04818.
PMC 2999987.
PMID 16738662.
^ Silva DA, Yu S, Ulge UY, Spangler JB, Jude KM, Labão-Almeida C, Ali LR, Quijano-Rubio A, Ruterbusch M, Leung I, Biary T, Crowley SJ, Marcos E, Walkey CD, Weitzner BD, Pardo-Avila F, Castellanos J, Carter L, Stewart L, Riddell SR, Pepper M, Bernardes GJ, Dougan M, Garcia KC, Baker D (January 2019).
"De novo design of potent and selective mimics of IL-2 and IL-15". Nature. 565 (7738): 186–191.
doi:
10.1038/s41586-018-0830-7.
PMC 6521699.
PMID 30626941.
^ "Rosetta's role in fighting coronavirus – Institute for Protein Design". Retrieved March 6, 2020.
^ Simons KT, Bonneau R, Ruczinski I, Baker D (1999). "Ab initio protein structure prediction of CASP III targets using Rosetta". Proteins. Suppl 3: 171–76.
doi:
10.1002/(SICI)1097-0134(1999)37:3+<171::AID-PROT21>3.0.CO;2-Z.
PMID 10526365.
^ "Interview with David Baker". Team Picard Distributed Computing. 2006. Archived from
the original on February 18, 2009. Retrieved December 23, 2008.
^ Nauli S, Kuhlman B, Baker D (July 2001). "Computer-based redesign of a protein folding pathway". Nature Structural Biology. 8 (7): 602–05.
doi:
10.1038/89638.
PMID 11427890.
^ Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D (November 2003). "Design of a novel globular protein fold with atomic-level accuracy". Science. 302 (5649): 1364–68.
Bibcode:
2003Sci...302.1364K.
doi:
10.1126/science.1089427.
PMID 14631033.
^ Jones DT (November 2003). "Structural biology. Learning to speak the language of proteins". Science. 302 (5649): 1347–48.
doi:
10.1126/science.1092492.
PMID 14631028.
^ von Grotthuss M, Wyrwicz LS, Pas J, Rychlewski L (June 2004). "Predicting protein structures accurately". Science. 304 (5677): 1597–99, author reply 1597–99.
doi:
10.1126/science.304.5677.1597b.
PMID 15192202.
^ "Articles citing: Kuhlman et al. (2003) 'Design of a novel globular protein fold with atomic-level accuracy'". ISI Web of Science. Retrieved July 10, 2008.
^ "October 2005 molecule of the month: Designer proteins". RCSB Protein Data Bank. Retrieved September 7, 2008.
^ "Kuhlman laboratory homepage". Kuhlman Laboratory. University of North Carolina. Retrieved September 7, 2008.
^ "RosettaDesign web server". Kuhlman Laboratory. University of North Carolina. Retrieved September 7, 2008.
^
Jump up to: a b Gray JJ, Moughon SE, Kortemme T, et al. (July 2003). "Protein–protein docking predictions for the CAPRI experiment". Proteins. 52 (1): 118–22.
CiteSeerX 10.1.1.80.9354.
doi:
10.1002/prot.10384.
PMID 12784377.
^ Daily MD, Masica D, Sivasubramanian A, Somarouthu S, Gray JJ (2005).
"CAPRI rounds 3–5 reveal promising successes and future challenges for RosettaDock". Proteins. 60 (2): 181–86.
CiteSeerX 10.1.1.521.9981.
doi:
10.1002/prot.20555.
PMID 15981262. Archived from
the original on June 30, 2012.
^ Méndez R, Leplae R, Lensink MF, Wodak SJ (2005).
"Assessment of CAPRI predictions in rounds 3–5 shows progress in docking procedures". Proteins. 60 (2): 150–69.
doi:
10.1002/prot.20551.
PMID 15981261. Archived from
the original on June 30, 2012.
^ "RosettaDock server". Rosetta Commons. Retrieved March 28, 2020.
^ "Protein–protein docking at Rosetta@home". Rosetta@home forums. University of Washington. Retrieved September 7, 2008.
^ "Robetta web server". Baker laboratory. University of Washington. Retrieved May 7, 2019.
^ Aloy P, Stark A, Hadley C, Russell RB (2003). "Predictions without templates: new folds, secondary structure, and contacts in CASP5". Proteins. 53 Suppl 6: 436–56.
doi:
10.1002/prot.10546.
PMID 14579333.
^ Tress M, Ezkurdia I, Graña O, López G, Valencia A (2005). "Assessment of predictions submitted for the CASP6 comparative modeling category". Proteins. 61 Suppl 7: 27–45.
doi:
10.1002/prot.20720.
PMID 16187345.
^ Battey JN, Kopp J, Bordoli L, Read RJ, Clarke ND, Schwede T (2007). "Automated server predictions in CASP7". Proteins. 69 Suppl 8: 68–82.
doi:
10.1002/prot.21761.
PMID 17894354.
^ Chivian D, Kim DE, Malmström L, Schonbrun J, Rohl CA, Baker D (2005). "Prediction of CASP6 structures using automated Robetta protocols". Proteins. 61 Suppl 7: 157–66.
doi:
10.1002/prot.20733.
PMID 16187358.
^ Baker D.
"David Baker's Rosetta@home journal, message 52902". Rosetta@home forums. University of Washington. Retrieved September 7, 2008.
^ Ovchinnikov, S; Kim, DE; Wang, RY; Liu, Y; DiMaio, F; Baker, D (September 2016).
"Improved de novo structure prediction in CASP11 by incorporating coevolution information into Rosetta". Proteins. 84 Suppl 1: 67–75.
doi:
10.1002/prot.24974.
PMC 5490371.
PMID 26677056.
^ Baker D.
"David Baker's Rosetta@home journal (message 52963)". Rosetta@home forums. University of Washington. Retrieved September 16, 2008.
^ "Foldit forums: How many users does Foldit have? Etc. (message 2)". University of Washington. Retrieved September 27, 2008.
^ "Foldit: Frequently Asked Questions". fold.it. University of Washington. Retrieved September 19,2008.
^ "Project list – BOINC". University of California. Retrieved September 8, 2008.
^ Pande Group (2010).
"High Performance FAQ".
Stanford University. Archived from
the original (FAQ) on September 21, 2012. Retrieved September 19, 2011.
^ 7im (April 2, 2010).
"Re: Answers to: Reasons for not using F@H". Retrieved September 19,2011.
^ Vijay Pande (August 5, 2011).
"Results page updated – new key result published in our work in Alzheimer's Disease". Retrieved September 19, 2011.
^ Pande Group.
"Folding@home Diseases Studied FAQ".
Stanford University. Archived from
the original (FAQ) on October 11, 2007. Retrieved September 12, 2011.
^ Vijay Pande (September 26, 2007).
"How FAH works: Molecular dynamics". Retrieved September 10, 2011.
^
Jump up to: a b tjlane (June 9, 2011).
"Re: Course grained Protein folding in under 10 minutes". Retrieved September 19, 2011.
^ jmn (July 29, 2011).
"Rosetta@home and Folding@home: additional projects". Retrieved September 19, 2011.
^ Pande Group.
"Client Statistics by OS". Stanford University. Retrieved October 18, 2011.
^
Jump up to: a b "Rosetta@home: Credit overview". boincstats.com. Retrieved March 28, 2020.
^ Malmström L, Riffle M, Strauss CE, et al. (April 2007).
"Superfamily assignments for the yeast proteome through integration of structure prediction with the gene ontology". PLOS Biology. 5 (4): e76.
doi:
10.1371/journal.pbio.0050076.
PMC 1828141.
PMID 17373854.
^ Bonneau R (2006).
"World Community Grid Message Board Posts: HPF -> HPF2 transition". Bonneau Lab, New York University. Retrieved September 7, 2008.
^ "List of Richard Bonneau's publications". Bonneau Lab, New York University. Archived from
the original on July 7, 2008. Retrieved September 7, 2008.
^ Bonneau R.
"World Community Grid Message Board Posts". Bonneau Lab, New York University. Archived from
the original on July 4, 2008. Retrieved September 7, 2008.
^ "Predictor@home: Developing new application areas for P@H". The Brooks Research Group. Retrieved September 7, 2008.[
dead link]
^ Carrillo-Tripp M (2007).
"dTASSER". The Scripps Research Institute. Archived from
the original on July 6, 2007. Retrieved September 7, 2008.
^ "RALPH@home website". RALPH@home forums. University of Washington. Retrieved September 7, 2008.
^ "Rosetta@home". Retrieved March 19, 2020.
^ "Rosetta@home: The new credit system explained". Rosetta@home forums. University of Washington. 2006. Retrieved October 8, 2008.
^ "BOINCstats: Project Credit Comparison". boincstats.com. 2008. Archived from
the originalon September 13, 2008. Retrieved October 8, 2008.
^ "Credit divided over projects". boincstats.com. Retrieved February 19, 2015.
^ "Rosetta@home: Predictor of the day archive". Rosetta@home. University of Washington. 2008. Archived from
the original on September 24, 2008. Retrieved October 8, 2008.
^ "Rosetta@home: Protein Folding, Design, and Docking". Rosetta@home. University of Washington. 2008. Retrieved October 8, 2008.
External links[
edit]
Official website
Baker Lab Baker Lab website
David Baker's Rosetta@home journal
BOINC Includes platform overview, and a guide to install BOINC and attach to Rosetta@home
BOINCstats – Rosetta@home Detailed contribution statistics
RALPH@home Website for Rosetta@home alpha testing project
Rosetta@home video on YouTube Overview of Rosetta@home given by David Baker and lab members
Rosetta Commons Academic collaborative for developing the Rosetta platform
Kuhlman lab webpage, home of RosettaDesign
Online Rosetta services
Rosetta Commons list of available servers
Robetta Protein structure prediction server
ROSIE Docking, design, etc. multifunctional server-set
RosettaDesign Protein design server
RosettaBackrub Flexible backbone / protein design server



